
Hemophilia for the USMLE Step 2
Start your One-Week Free Trial
Already subscribed? Log in »
Hemophilia for the USMLE Step 2 Exam
- Definition:
- Hemophilia is an X-linked recessive bleeding disorder marked by a deficiency in specific clotting factors, leading to impaired coagulation.
- Primarily affects males, while females can be carriers, with rare symptomatic cases in females due to skewed X-chromosome inactivation.
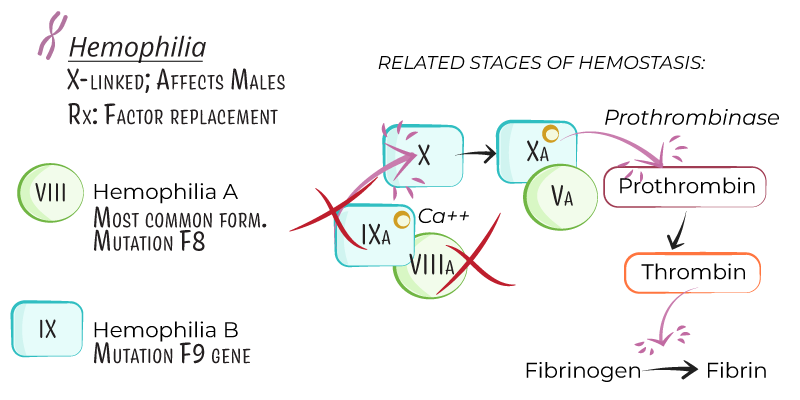
- Types:
- Hemophilia A: Factor VIII deficiency, accounting for ~80% of cases.
- Hemophilia B (Christmas Disease): Factor IX deficiency, comprising ~20% of cases.
Genetics and Pathophysiology
- Inheritance:
- Hemophilia A and B are both X-linked recessive, affecting mostly males.
- Carrier females have a 50% chance of passing the affected gene to sons (affected) or daughters (carriers).
- Pathogenesis:
- Factor VIII (Hemophilia A) and Factor IX (Hemophilia B) are part of the intrinsic coagulation pathway and are necessary to activate factor X, which leads to thrombin generation and fibrin clot formation.
- Deficiency disrupts this pathway, prolonging bleeding and impairing stable clot formation.

- Severity:
- Severe: Factor level <1% of normal, often with spontaneous bleeding.
- Moderate: Factor level 1–5% of normal, with bleeding after mild trauma.
- Mild: Factor level 5–40% of normal, with bleeding typically only after significant trauma or surgery.
Clinical Presentation
- Bleeding Sites:
- Hemarthrosis: Repeated bleeding into joints (knees, elbows, ankles), leading to joint deformity and hemophilic arthropathy if untreated.
- Muscle Bleeding: Deep muscle bleeding, particularly in the thigh or iliopsoas, leading to compartment syndrome if not managed.
- Mucocutaneous Bleeding: Less common but may occur, especially during dental procedures.
- Intracranial Hemorrhage: Rare but life-threatening, often from trauma, even minor head injuries.
- Post-Surgical or Post-Traumatic Bleeding:
- Prolonged bleeding after procedures or minor trauma, particularly in mild or moderate hemophilia, may delay diagnosis.
Diagnosis
- Laboratory Tests:
- CBC: Typically normal, though may show anemia with significant blood loss.
- Prothrombin Time (PT): Normal, as PT measures the extrinsic pathway, unaffected by factor VIII or IX deficiencies.
- Activated Partial Thromboplastin Time (aPTT): Prolonged, as aPTT evaluates the intrinsic pathway.
- Specific Factor Assays: Factor VIII and IX levels confirm diagnosis and determine severity.
- Genetic Testing:
- Useful for carrier detection and prenatal diagnosis in families with a known history of hemophilia.
- Inhibitor Screening:
- Antibodies to factor VIII or IX can develop in some patients, especially in severe hemophilia A, complicating treatment.
Treatment
- Factor Replacement Therapy:
- Factor VIII (Hemophilia A) and Factor IX (Hemophilia B):
- On-Demand Therapy: Used during active bleeding episodes.
- Prophylactic Therapy: Regular infusions to maintain factor levels and prevent bleeding, especially in severe cases.
- Dosing: Based on body weight, target level, and bleeding type.
- Bypassing Agents for Inhibitors:
- Recombinant Factor VIIa (rFVIIa) and Activated Prothrombin Complex Concentrate (aPCC): Used in patients with inhibitors against factor VIII or IX.
- Emicizumab:
- Mechanism: A bispecific antibody mimicking factor VIII function, allowing effective clotting in hemophilia A patients, even with inhibitors.
- Administration: Subcutaneous, typically weekly or biweekly for prophylaxis.
- Adjunctive Therapies:
- Antifibrinolytics (e.g., tranexamic acid): Useful for mucosal bleeding, such as in dental procedures.
- Pain Management: Acetaminophen preferred; NSAIDs are avoided due to platelet inhibition.
Key Points
- Hemophilia is an X-linked recessive disorder due to deficiencies in factor VIII (hemophilia A) or factor IX (hemophilia B), primarily affecting males.
- Severity of hemophilia is based on factor activity, with severe cases often presenting with spontaneous joint and muscle bleeding.
- Diagnosis includes prolonged aPTT, specific factor assays, and genetic testing to identify carriers.
- Management includes on-demand and prophylactic factor replacement therapy, with bypassing agents for inhibitor patients and emicizumab for prophylaxis in hemophilia A.
- Complications include joint deformities from recurrent hemarthrosis, inhibitor development, and risk of bleeding in trauma or surgery.