
USMLE/COMLEX 2 - Adrenal Insufficiency
Start your One-Week Free Trial
Already subscribed? Log in »
Here are key facts for USMLE Step 2 & COMLEX-USA Level 2 from the Adrenal Insufficiency tutorial, as well as points of interest at the end of this document that are not directly addressed in this tutorial but should help you prepare for the boards. See the tutorial notes for further details and relevant links.
- --
VITAL FOR USMLE/COMLEX 2
Clinical Presentation and Diagnosis
1. Be suspicious of adrenal crisis in patients with acute shock that is refractory to vasopressors and fluid replacement.
2. In chronic AI, adrenal reserves may initially maintain basal hormone levels, but with impaired ACTH stress response.
3. Primary AI patients present with hyperpigmentation of the skin and mucosa (gums and hand folds), hypotension, hyponatremia, hyperkalemia, and metabolic acidosis.
4. Symptoms of chronic PAI include weakness, fatigue, weight loss, gastrointestinal problems, and possibly salt cravings.
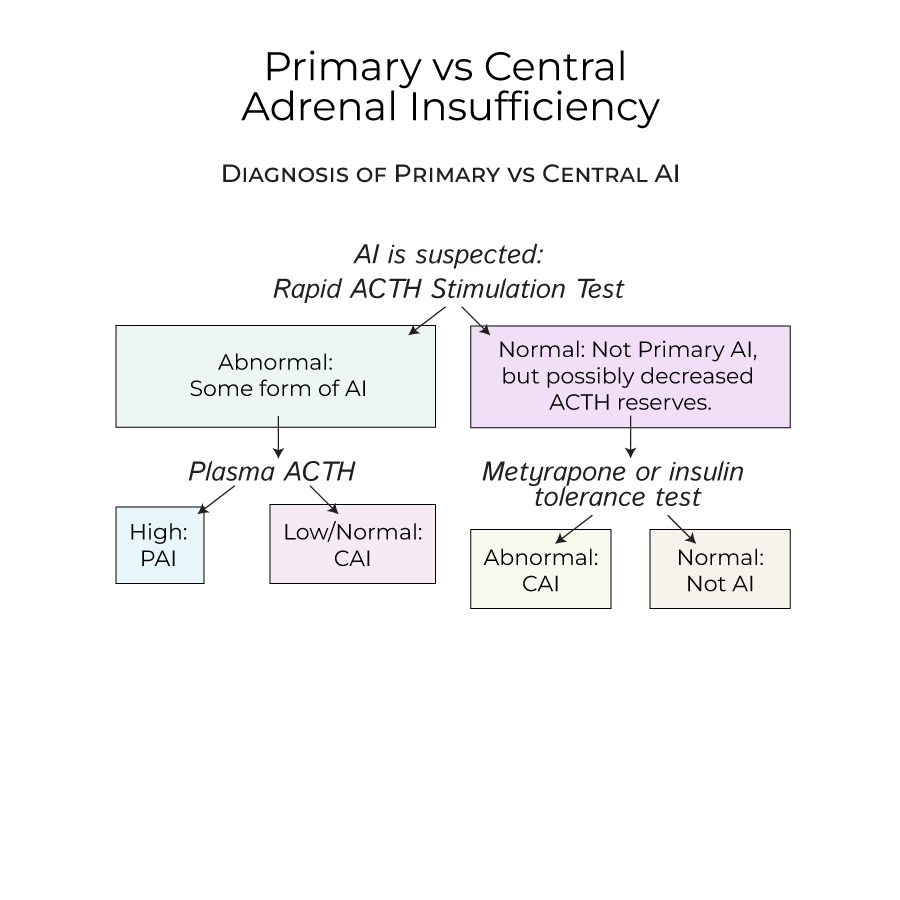
5. When AI is suspected, perform a rapid ACTH stimulation test, which raises cortisol levels in healthy individuals.
Adrenal Crisis Management
1. Acute adrenal crisis is a life-threatening situation with shock, fever, dehydration, nausea, vomiting, hypoglycemia, apathy, and weakness.
2. Adrenal crisis occurs when patients with adrenal insufficiency face additional stressors (infections, trauma, surgery, dehydration).
3. To avoid acute adrenal crisis, provide additional doses of glucocorticoids when stressors, such as surgery, are expected.
4. Patients can carry medical cards that warn of their condition in case of accidental trauma.
Treatment Principles
1. Treatment for AI includes glucocorticoid replacement for all forms and mineralocorticoid replacement for primary AI.
2. Beware of overtreatment with glucocorticoids, as excessive cortisol exposure produces Cushing's syndrome.
3. Adrenal crisis is a medical emergency requiring immediate intervention.
- --
HIGH YIELD
Types of Adrenal Insufficiency
1. Primary adrenal insufficiency affects slightly more women than men and is typically diagnosed in patients 30-50 years old.
2. Central adrenal insufficiency primarily affects cortisol and androgen secretion, with preserved aldosterone secretion.
3. Central AI is more common than Primary AI.
Causes of Primary AI
1. Autoimmune diseases are the most common causes of primary AI in the United States, with anti-adrenal antibodies destroying the adrenal cortex.
2. Autoimmune Polyendocrine Syndrome Type 1 (APS-1) typically onset in childhood with adrenal insufficiency, hypoparathyroidism, and mucocutaneous candidiasis.
3. Recurrent mucocutaneous candidiasis in children is a potential indicator of APS-1, manifesting as thrush and angular cheilitis.
4. Autoimmune Polyendocrine Syndrome Type 2 (APS-2) is associated with adrenal insufficiency, Hashimoto's thyroiditis, and Type I diabetes mellitus.
5. Infections are important causes of Primary AI, particularly in countries where TB and/or HIV are endemic.
6. Bilateral adrenal hemorrhage is a cause of acute PAI, with the adrenal gland vulnerable due to its blood supply (three arteries, one vein).
7. Waterhouse-Friderichsen Syndrome in children presents with adrenal bleeding caused by septicemia, most often associated with meningococcal or pseudomonas infections.
Causes of Central AI
1. Most commonly caused by long-term exogenous steroid use, developing from sudden cessation or inability of the HPA axis to respond to stressors.
2. May be caused by dysfunction in the pituitary or hypothalamus, including tumors, infections, and drugs.
3. Drugs that interrupt ACTH production include immune checkpoint inhibitors, high-dose progestins, and opioids.
4. Pituitary and/or hypothalamic dysfunction is often associated with deficiencies of other pituitary hormones.
Differential Diagnosis
1. Important clinical distinctions between PAI and CAI:
- No hyperpigmentation in central AI (ACTH is deficient)
- Less hypovolemia or hypotension in central AI (aldosterone secretion is normal)
- --
Beyond the Tutorial
Clinical Pearls for Management
1. Consider adrenal insufficiency in patients with unexplained hypotension, especially when resistant to conventional treatments.
2. Check baseline morning cortisol and ACTH levels before initiating steroid therapy when possible.
3. Adjust glucocorticoid replacement during illness using the "sick day rules" - typically doubling or tripling the usual dose.
4. Monitor for signs of overreplacement: weight gain, hypertension, edema, and glucose intolerance.
Emerging Concepts
1. Continuous subcutaneous hydrocortisone infusion may better mimic physiological cortisol secretion.
2. Delayed-release hydrocortisone formulations can improve quality of life and metabolic parameters.
3. Regular screening for cardiovascular risk factors is important in patients on long-term glucocorticoid therapy.
4. Consider osteoporosis prophylaxis for patients on chronic glucocorticoid therapy.
Complications and Prognosis
1. Even with appropriate therapy, patients with adrenal insufficiency have increased mortality rates.
2. Poor adherence to replacement therapy is a common cause of recurrent hospitalizations.
3. Long-term follow-up should include assessment for other autoimmune conditions in patients with primary AI.
4. Quality of life may remain reduced even with adequate hormone replacement.