
Interstitial Lung Diseases for ABIM
Start your One-Week Free Trial
Already subscribed? Log in »
Interstitial Lung Diseases for the American Board of Internal Medicine Exam
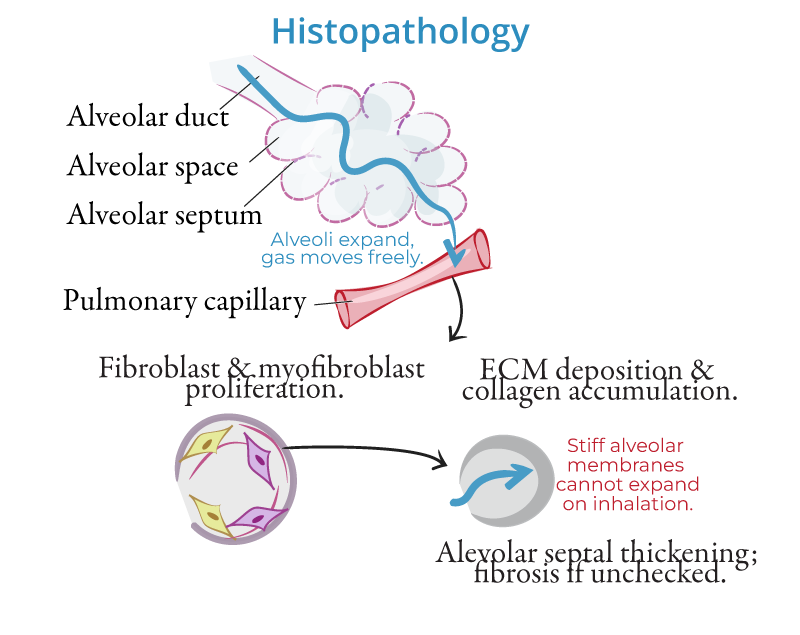
Pathophysiology of Interstitial Lung Disease (ILD)

- Chronic Inflammation and Fibrosis:
- Interstitial lung diseases (ILDs) are a group of disorders characterized by chronic inflammation and scarring (fibrosis) of the lung interstitium, the tissue surrounding the alveoli. The inflammatory process causes thickening of the alveolar walls, limiting gas exchange and leading to progressive respiratory insufficiency.
- Fibrotic Changes:
- Persistent injury to the lung tissue results in abnormal healing and fibroblast activation, which lays down excessive collagen, resulting in fibrosis. This disrupts the normal architecture of the lungs, causing restrictive lung disease.
- Mechanisms of Damage:
- Repeated exposure to harmful agents such as toxins, dust, drugs, or infections can cause chronic damage to the lung parenchyma. In some cases, autoimmune mechanisms play a role, leading to persistent inflammation and fibrosis.
Etiologies
- Idiopathic Pulmonary Fibrosis (IPF):
- A chronic, progressive form of ILD with no known cause. IPF is associated with abnormal wound healing after lung injury, leading to fibrosis.
- Pneumoconioses:
- Caused by inhalation of occupational dusts:
- Asbestosis: From asbestos exposure, common in construction and shipbuilding industries.
- Silicosis: From inhaling silica dust, often seen in miners or stone workers.
- Coal Workers' Pneumoconiosis: Black lung disease caused by inhaling coal dust.
- Hypersensitivity Pneumonitis (HP):
- An immune-mediated ILD triggered by inhalation of organic dusts, such as bird droppings, moldy hay, or contaminated humidifiers.
- Sarcoidosis:
- A multisystem granulomatous disorder that frequently affects the lungs. Granulomas form in the lung parenchyma, leading to fibrosis in chronic cases.
- Connective Tissue Diseases:
- ILD can be associated with systemic autoimmune disorders, including:
- Rheumatoid arthritis (RA): May lead to fibrosis in the lungs.
- Systemic sclerosis (scleroderma): Causes diffuse fibrosis.
- Polymyositis/Dermatomyositis: Can lead to interstitial lung involvement.
- Drug-Induced ILD:
- Medications such as amiodarone, bleomycin, and methotrexate can cause lung toxicity, leading to inflammation and fibrosis.
Clinical Features
- Dyspnea:
- Progressive shortness of breath is the most common symptom, typically worsening over months to years.
- Dry Cough:
- A nonproductive cough that is often persistent.
- Inspiratory Crackles:
- Fine, velcro-like crackles heard on auscultation, particularly at the lung bases, are a hallmark of ILD.
- Digital Clubbing:
- Seen in more advanced disease, particularly in IPF and asbestosis.
- Extrapulmonary Manifestations:
- Sarcoidosis may present with lymphadenopathy, skin lesions, and uveitis. Connective tissue disease-related ILD may have joint pain, skin thickening, or muscle weakness.
Diagnosis
- High-Resolution CT (HRCT):
- The gold standard imaging modality for ILD. Common findings include:
- Ground-glass opacities: Indicating active inflammation.
- Honeycombing: Represents advanced fibrosis and is typically seen in idiopathic pulmonary fibrosis.
- Reticulations: A mesh-like appearance of fibrosis.
- Pulmonary Function Tests (PFTs):
- Show a restrictive pattern with decreased total lung capacity (TLC) and forced vital capacity (FVC). Diffusion capacity for carbon monoxide (DLCO) is also reduced, indicating impaired gas exchange.
- Bronchoalveolar Lavage (BAL):
- Fluid from the lungs can be analyzed to identify eosinophils, lymphocytes, or neutrophils, depending on the type of ILD. This is particularly useful in diagnosing hypersensitivity pneumonitis or sarcoidosis.
- Lung Biopsy:
- In cases where the diagnosis is unclear, a surgical biopsy may be required to confirm the type of ILD, especially in suspected cases of IPF or drug-induced ILD.
- Serologic Testing:
- Blood tests for autoimmune markers such as rheumatoid factor, antinuclear antibodies (ANA), and anti-cyclic citrullinated peptide (anti-CCP) can help diagnose connective tissue disease-associated ILD.
Management
General Approach
- Smoking Cessation:
- Smoking worsens lung fibrosis and increases the risk of lung cancer in patients with ILD, particularly those exposed to asbestos.
- Avoidance of Triggers:
- Patients with hypersensitivity pneumonitis should avoid exposure to known antigens (e.g., birds, mold) that trigger lung inflammation.
- Supplemental Oxygen:
- Many patients with advanced ILD require oxygen therapy to maintain adequate oxygen saturation levels, particularly during exertion or sleep.
Pharmacologic Therapy
- Idiopathic Pulmonary Fibrosis (IPF):
- Antifibrotic agents such as pirfenidone and nintedanib slow the progression of fibrosis in IPF but do not reverse the disease. They reduce the decline in lung function and may improve survival.
- Steroids and Immunosuppressants:
- Corticosteroids (e.g., prednisone) are used in sarcoidosis, hypersensitivity pneumonitis, and autoimmune-associated ILD. In cases where steroids are insufficient, methotrexate, azathioprine, or mycophenolate mofetil may be added.
- Antibiotics:
- Long-term antibiotics (e.g., doxycycline or macrolides) may be considered in certain cases of hypersensitivity pneumonitis or organizing pneumonia.
Surgical Therapy
- Lung Transplantation:
- For patients with advanced ILD and respiratory failure, lung transplantation offers the only definitive treatment. Candidates are selected based on disease progression, age, and comorbidities.
Supportive Care
- Pulmonary Rehabilitation:
- Exercise training, education, and breathing strategies improve functional capacity and quality of life in ILD patients.
- Vaccinations:
- Annual influenza and pneumococcal vaccinations are recommended to prevent respiratory infections, which can worsen ILD.
Prognosis
- Idiopathic Pulmonary Fibrosis (IPF):
- IPF has the worst prognosis among ILDs, with a median survival of 3 to 5 years after diagnosis. Disease progression is often rapid, leading to respiratory failure.
- Hypersensitivity Pneumonitis:
- Prognosis depends on early recognition and antigen avoidance. Chronic exposure leads to progressive fibrosis and irreversible lung damage.
- Sarcoidosis:
- Many cases resolve spontaneously or respond to corticosteroids. However, chronic pulmonary involvement can lead to fibrosis and respiratory failure in severe cases.
Key Points
- Interstitial lung diseases (ILDs) involve chronic inflammation and fibrosis of the lung interstitium, impairing gas exchange and causing progressive dyspnea and cough.
- Etiologies include idiopathic pulmonary fibrosis (IPF), pneumoconioses (asbestosis, silicosis), hypersensitivity pneumonitis, sarcoidosis, connective tissue disease-associated ILD, and drug-induced ILD.
- Diagnosis relies on high-resolution CT (HRCT), showing patterns such as honeycombing and ground-glass opacities, and pulmonary function tests, which reveal a restrictive pattern.
- Management includes smoking cessation, avoiding triggers, antifibrotic agents (for IPF), corticosteroids, and immunosuppressants for autoimmune and inflammatory causes.
- Lung transplantation is an option for advanced disease, and pulmonary rehabilitation is beneficial for maintaining function and quality of life.
- Prognosis varies by cause, with idiopathic pulmonary fibrosis having the poorest outcomes, while early detection and management improve outcomes in other forms of ILD.