
Hemophilia for the ABIM
Start your One-Week Free Trial
Already subscribed? Log in »
Hemophilia for the American Board of Internal Medicine Exam
Overview of Hemophilia
- Definition:
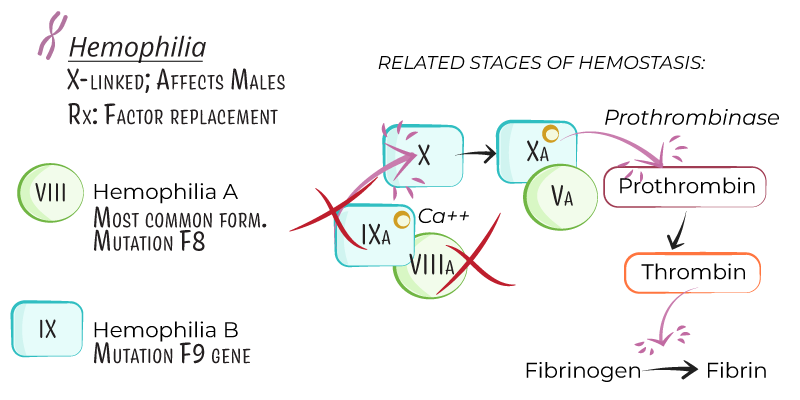
- Hemophilia is an X-linked recessive bleeding disorder characterized by a deficiency in specific clotting factors, leading to impaired blood coagulation.
- Hemophilia primarily affects males, with females as carriers, although symptomatic females can arise from lyonization (X-chromosome inactivation).
- Types:
- Hemophilia A: Caused by a deficiency in factor VIII, accounting for approximately 80% of cases.
- Hemophilia B (Christmas Disease): Caused by a deficiency in factor IX and constitutes around 20% of cases.
Genetics and Pathophysiology
- Inheritance:
- Hemophilia A and B are X-linked recessive, predominantly affecting males.
- Female carriers have a 50% chance of passing the gene to sons (affected) and daughters (carriers).
- Pathogenesis:
- Factor VIII (Hemophilia A) and Factor IX (Hemophilia B) are intrinsic pathway proteins that activate factor X, leading to thrombin formation and fibrin clot development.
- Deficiency of these factors disrupts the clotting cascade, prolonging bleeding.

- Severity:
- Severe: Factor level <1% of normal, associated with spontaneous bleeding.
- Moderate: Factor level 1–5% of normal, with bleeding following minor trauma.
- Mild: Factor level 5–40% of normal, with bleeding only after significant trauma or surgery.
Clinical Presentation
- Bleeding Sites:
- Hemarthrosis: Bleeding into joints, especially knees, elbows, and ankles, is the hallmark of severe hemophilia. Recurrent hemarthrosis can lead to joint deformity and arthropathy.
- Muscle Bleeding: Deep muscle bleeding, particularly in the thigh, calf, and iliopsoas, leading to compartment syndrome if untreated.
- Mucocutaneous Bleeding: Less common in hemophilia than in von Willebrand’s disease; bleeding may occur with dental procedures or nasal trauma.
- Intracranial Hemorrhage: Rare but life-threatening, often following trauma, even minor head injuries.
- Other Presentations:
- Postoperative or Post-Traumatic Bleeding: Extended bleeding after procedures or injuries, especially in mild or moderate hemophilia, where diagnosis may be delayed.
Diagnosis of Hemophilia
- Laboratory Tests:
- Complete Blood Count (CBC): Typically normal; may show anemia if there is significant blood loss.
- Prothrombin Time (PT): Normal, as PT measures the extrinsic pathway, unaffected by factor VIII or IX deficiencies.
- Activated Partial Thromboplastin Time (aPTT): Prolonged, as aPTT assesses the intrinsic pathway. Normal aPTT does not exclude mild hemophilia.
- Factor Assays:
- Specific factor VIII and IX activity levels confirm the diagnosis and determine the severity of hemophilia.
- Genetic Testing:
- Useful for carrier detection and prenatal diagnosis, especially in families with known hemophilia.
- Inhibitor Screening:
- Inhibitors: Antibodies against infused factor VIII or IX may develop, especially in severe hemophilia A, complicating treatment.
- Bethesda Assay: Measures inhibitor titer, with levels >5 Bethesda units (BU) classified as high-titer inhibitors, requiring alternative treatment approaches.
Management of Hemophilia
- General Measures:
- Avoid medications that affect platelet function (e.g., aspirin, NSAIDs).
- Encourage regular physical activity to strengthen muscles around joints, reducing bleeding risk, and prevent joint deformities.
- Immunize for hepatitis A and B to reduce infection risk, as factor concentrates can carry risk of bloodborne pathogens.
- Factor Replacement Therapy:
- Factor VIII Concentrate (for Hemophilia A) and Factor IX Concentrate (for Hemophilia B):
- On-Demand Therapy: Administered during bleeding episodes.
- Prophylactic Therapy: Regular infusions to maintain factor levels and prevent bleeding, especially in severe hemophilia.
- Dosing: Calculated based on factor deficiency level, body weight, and type of bleeding or surgery planned.
- Extended Half-Life (EHL) Products:
- Modified factor VIII or IX concentrates with extended half-lives, allowing less frequent dosing in prophylaxis, increasing patient compliance.
- Inhibitor Management:
- Bypassing Agents:
- Recombinant Factor VIIa (rFVIIa): Activates factor X without needing factors VIII or IX, effective in high-titer inhibitor patients.
- Activated Prothrombin Complex Concentrate (aPCC): Contains activated clotting factors, also used in inhibitor patients.
- Immune Tolerance Induction (ITI):
- Regular high-dose factor infusions to induce immune tolerance in inhibitor patients, particularly useful in young children.
- Emicizumab:
- Mechanism: A bispecific antibody that mimics factor VIII function by bridging factors IXa and X, effective in hemophilia A with or without inhibitors.
- Administration: Subcutaneous dosing, usually weekly or every other week, effective for both prophylaxis and bleeding control.
- Adjunctive Therapies:
- Antifibrinolytics (e.g., tranexamic acid, aminocaproic acid): Stabilize clots in mucosal bleeding, useful in dental procedures or minor surgeries.
- Pain Management: NSAIDs are generally avoided; acetaminophen or opioids are preferred for pain from hemarthrosis or muscle bleeding.
Complications of Hemophilia
- Joint Deformity and Hemophilic Arthropathy:
- Repeated hemarthrosis causes synovial inflammation, cartilage damage, and chronic pain, leading to joint deformities and reduced mobility.
- Inhibitor Development:
- Inhibitors to factor VIII or IX are more common in hemophilia A, complicating treatment due to resistance to standard factor replacement.
- Infectious Risks:
- While modern factor products are highly purified, older treatments posed a risk for bloodborne pathogens (e.g., HIV, hepatitis B, and C).
- Psychosocial Impact:
- The chronic nature of hemophilia and the need for frequent infusions impact quality of life, leading to psychosocial challenges and depression in some patients.
Key Points
- Hemophilia is an X-linked recessive bleeding disorder due to deficiencies in factor VIII (hemophilia A) or factor IX (hemophilia B), impairing the intrinsic coagulation pathway.
- Classification by severity (severe, moderate, mild) depends on factor levels, with severe hemophilia often presenting with spontaneous joint and muscle bleeding.
- Diagnosis includes prolonged aPTT and specific factor assays, with genetic testing for carrier and prenatal diagnosis.
- Treatment includes on-demand or prophylactic factor replacement therapy, with emicizumab and bypassing agents for patients with inhibitors.
- Complications include joint deformities from hemarthrosis, inhibitor development, and potential psychosocial impacts due to the chronic nature of the disease and need for lifelong management.